De nouveaux mécanismes pathologiques du syndrome de l'atrophie optique humaine dévoilés
- Recherche
le 4 juillet 2019
L’équipe de Michèle Studer, Directrice de Recherche Inserm à l’Institut de Biologie Valrose, en collaboration avec deux équipes des universités de Madrid et Séville, vient de générer un modèle de souris mutantes qui recrée le syndrome de Bosch-Boonstra-Schaaf (BBSOAS), maladie neuro-développementale qui associe une atrophie optique à une déficience intellectuelle. Ces travaux sont publiés le 18 juillet dans la revue EMBO Molecular Medicine.
Le syndrome de Bosch-Boonstra-Schaaf (BBSOAS), maladie neuro-développementale qui associe une atrophie optique à une déficience intellectuelle, est causé chez l’homme par la mutation du gène NR2F1 (#615722 dans la base de données des maladies génétique OMIM). L’équipe de Michèle Studer, Directrice de Recherche Inserm à l’Institut de Biologie Valrose, en collaboration avec deux équipes des universités de Madrid et Séville, vient de générer un modèle de souris mutantes qui recrée cette maladie. Ces travaux qui seront prochainement publiés dans la revue EMBO Molecular Medicine, ont permis de déchiffrer les mécanismes cellulaires et moléculaires à l’origine de l’atrophie oculaire et des problèmes visuels induits par le BBSOAS et proposent une approche thérapeutique potentielle qui pourrait s’appliquer à d’autres neuropathies optiques de type génétique.
Une centaine d’enfants au monde sont affectés par l'atrophie optique de Bosch-Boonstra-Schaaf (BBSOA) présentant un éventail variable de déficits cliniques, visuels et cognitifs. Une association, créée aux États- Unis, a permis de regrouper les familles des patients permettant aux scientifiques et aux cliniciens de collecter des données génétiques et cliniques. A ce stade, nous sommes encore loin de proposer une possible thérapie. Cependant, les généticiens ont récemment pu relier le syndrome BBSOA à des mutations dans le gène NR2F1, gène clé du développement neuronal. Du fait de son extrême rareté, très peu d’informations sur l’origine de cette maladie sont à ce jour disponibles avec un faible espoir d’avancées à court terme. Pourtant, au vu des récents résultats de l’équipe de recherche dirigée par le Dr Michèle Studer à l’iBV à Nice, qui s’est intéressée à la caractérisation des mécanismes responsables de la physiopathologie de cette maladie génétique, un espoir s’est ouvert.
L’idée gagnante de l’équipe a été de générer un modèle de souris mutant dans lequel le gène analogue à celui des patients, très conservé entre l’homme et la souris, est génétiquement modifié. Le but : recréer au laboratoire un modèle de la maladie pour disséquer les mécanismes cellulaires et moléculaires qui mènent à une baisse progressive de l’acuité visuelle chez les enfants affectés. C’est avec cette idée que le Dr Michel Bertacchi, premier signataire de l’étude publiée, en collaboration avec l’équipe du Dr Paola Bovolenta à Madrid (spécialiste de l’étude de la rétine des vertébrés), a étudié le développement du système visuel dans leur modèle de souris. Les chercheurs se sont focalisés sur le nerf optique reliant la rétine aux autres structures cérébrales importantes pour la vision, tel que le cortex visuel. Les résultats montrent un scénario beaucoup plus compliqué que ce que l’on pouvait imaginer initialement.
Plusieurs facteurs agissent en parallèle : un retard de la différenciation des neurones de la rétine formant le nerf optique, appelés cellules ganglionnaires, et une inflammation chronique des cellules responsables du maintien structural des fibres axonales, les astrocytes. Tous ces facteurs convergent vers un résultat unique : la dégénérescence ou atrophie du nerf optique chez les souris mutantes pour Nr2f1, comme cela est observé chez les jeunes patients. C’est exactement ce qu’on appelle une neuropathie optique.
Parallèlement, grâce à la collaboration avec l’équipe dirigée par le Dr J.M. Delgado-Garcia à Séville, les chercheurs ont pu observer une diminution de la conduction des stimuli électriques le long du nerf optique. Leurs expériences montrent que ce défaut est dû à un dysfonctionnement des oligodendrocytes. Dans les conditions physiologiques normales, ces cellules spécialisées sont censées entourer les fibres axonales avec une gaine de myéline permettant une conduction électrique optimale ; dans la maladie, cette gaine de myéline semble donc affectée. C’est en effet ce que les chercheurs ont pu finalement démontrer sur leur modèle souris en injectant le Miconazole, composé pharmaceutique qui active la prolifération et la différenciation des oligodendrocytes. Le niveau de production de myéline est alors partiellement restauré. Reste à savoir si ce traitement sera validé comme efficace chez l’homme.
Cette avancée importante avec l’obtention d’un modèle animal porteur de cette maladie neuro- développementale, qui affecte également le bon fonctionnement du cerveau, a permis d’envisager le développement de nouveaux traitements améliorant les neuropathies optiques et ouvrant une voie prometteuse pour de futures approches thérapeutiques.
- Titre : Mouse Nr2f1 haploinsufficiency unveils new pathological mechanisms of a human optic atrophy syndrome
- Auteurs : Michele Bertacchi (1,2), Agnès Gruart (3), Polynikis Kaimakis (4,5), Cécile Allet (6,7), Linda Serra (1,8), Paolo Giacobini (6,7), José M. Delgado-García (3), Paola Bovolenta (4,5) and Michèle Studer (1)
- 1 - iBV, Institut de Biologie Valrose, Université Côte d’Azur - Inserm - CNRS, Nice, France
- 2 Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italie
- 3 Division of Neurosciences, Pablo de Olavide University, 12 Seville, Espagne
- 4 Centro de Biología Molecular “Severo Ochoa”, CSIC-UAM
- 5 CIBER de 13 Enfermedades Raras (CIBERER), Campus de la Universidad Autónoma de Madrid, Espagne
- 6 Jean-Pierre Aubert Research Center (JPArc), Laboratory of Development and Plasticity of the Neuroendocrine
Brain, Inserm, UMR-S 1172, Lille
- 7 University of Lille, FHU 1,000 Days 16 for Health, Lille
- 8 Department of Biotechnology and Biological Sciences, University of 17 Milano-Bicocca, Milano, Italie

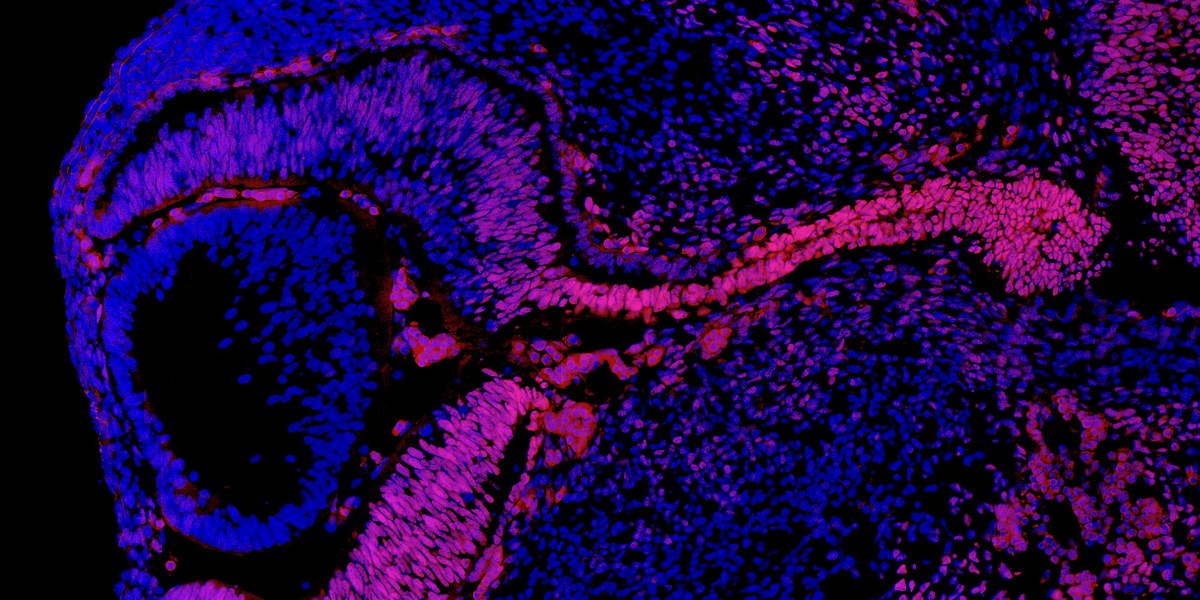
Pendant le développement embryonnaire de la souris, le gène Nr2f1 (en rouge) s’exprime dans la rétine (à gauche) ainsi que dans le nerf optique (à droite), qui relie l’œil au cerveau. Les chercheurs ont montré que l’inactivation de ce gène chez la souris provoque une atrophie optique qui ressemble à celle identifiée chez les patients porteurs de mutations du gène NR2F1.


